Uredba EU za medicinske pripomočke (2017/745)
Uredba (EU) 2017/745
Nova Uredba (EU) za medicinske pripomočke 2017/745 (MDR) je bila objavljena v Uradnem listu Evropske unije 5. maja 2017, in začela veljati 25. maja 2017. MDR bo nadomestila Direktivo o medicinskih pripomočkih (MDD) 93/42/EEC in Direktivo o aktivnih implantabilnih medicinskih pripomočkih (AIMD) 90/385 /EGS. SIQ Ljubljana je v postopku pridobitve imenovanja po Uredbi (EU) 2017/745. Certifikati, ki so bili v skladu z MDD izdani po objavi MDR (25. maj 2017), ostanejo veljavni do konca obdobja, navedenega na certifikatu, ki ne presega pet let od datuma njegove izdaje. Vendar postanejo neveljavni najpozneje 27. maja 2024. 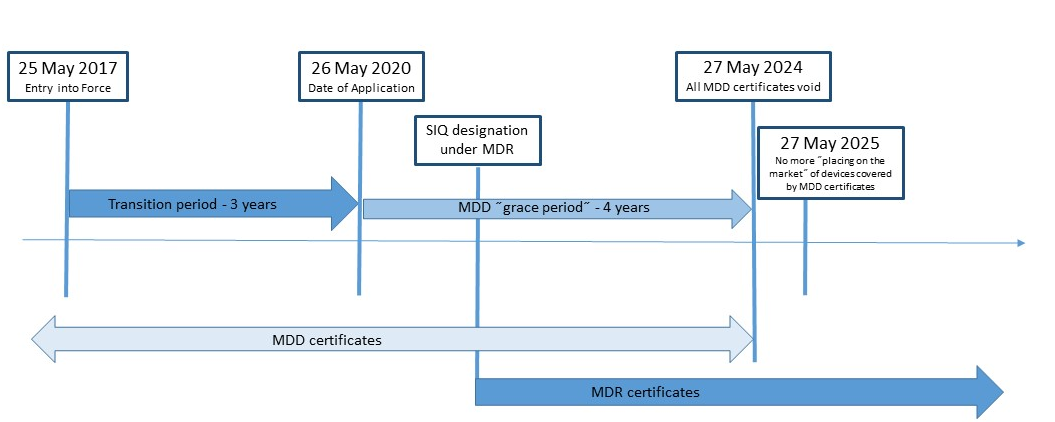
Zakaj pridobiti certifikat
Zakonodaja EU (Uredba MDR) o medicinskih pripomočkih določa, da morajo proizvajalci medicinskih pripomočkov svoje proizvode označiti s CE oznako, preden jih ponudijo na trg EU. S tem izjavljajo, da je medicinski pripomoček skladen s predpisi EU, ki urejajo področje medicinskih pripomočkov ter zagotavljajo, da je njihov medicinski pripomoček varen in strokovno ustrezen. Kadar je medicinski pripomoček razreda Is, Im, Ir, IIa, IIb in III (MDR), je potrebno v postopek ugotavljanja skladnosti medicinskega pripomočka vključiti tudi priglašeni organ (SIQ). Skladnost z zahtevami predpisov EU, ki urejajo medicinske pripomočke, se izkazuje s podeljenim EU certifikatom (EU Certificate).
Sistem vodenja kakovosti po ISO 13485 in Uredba (EU) 2017/745
ISO 13485 je harmonizirani standard, s katerim proizvajalci medicinskih pripomočkov dokazujejo usklajenost sistema kakovosti skladno z zahtevami uredbe MDR. Poleg zahtev standarda pa morajo proizvajalci vključiti tudi posebne zahteve, ki jih določa uredba. Pri postopkih ocenjevanja skladnosti po Aneksu IX in Aneksu XI del A, priporočamo, da ima proizvajalec že vzpostavljen sistem kakovosti po standardu ISO 13485.
Certifikacija medicinskih pripomočkov po Uredbi (EU) 2017/745
Medicinski pripomočki se lahko ponudijo na trg EU in nosijo oznako CE, če izpolnjujejo bistvene zahteve uredbe (MDR). Medicinski pripomočki se glede na stopnjo tveganja razvrščajo v:
- razred I – medicinski pripomočki z nizko stopnjo tveganja za uporabnika,
- razred IIa– medicinski pripomočki z večjo stopnjo tveganja za uporabnika,
- razred IIb– medicinski pripomočki z visoko stopnjo tveganja za uporabnika in
- razred III– medicinski pripomočki z najvišjo stopnjo tveganja za uporabnika.
Postopek certifikacije sestavljata presoja dokumentacije/tehnične mape medicinskega pripomočka in certifikacijska presoja. Priporočeno je, da ima proizvajalec medicinskih pripomočkov vzpostavljen sistem kakovosti po standardu ISO 13485. Presoja po medicinski uredbi se lahko kombinira s presojo po standardu ISO 13485. Po podelitvi EU certifikata in certifikata po ISO 13485 enkrat letno, z rednimi presojami v posameznih delih sistema, preverjamo delovanje sistema, enkrat v treh letih pa izvedemo obnovitveno presojo. EU certifikat je veljaven, dokler z vsakoletnimi presojami dokazujete, da izpolnjujete zahteve ISO 13485. 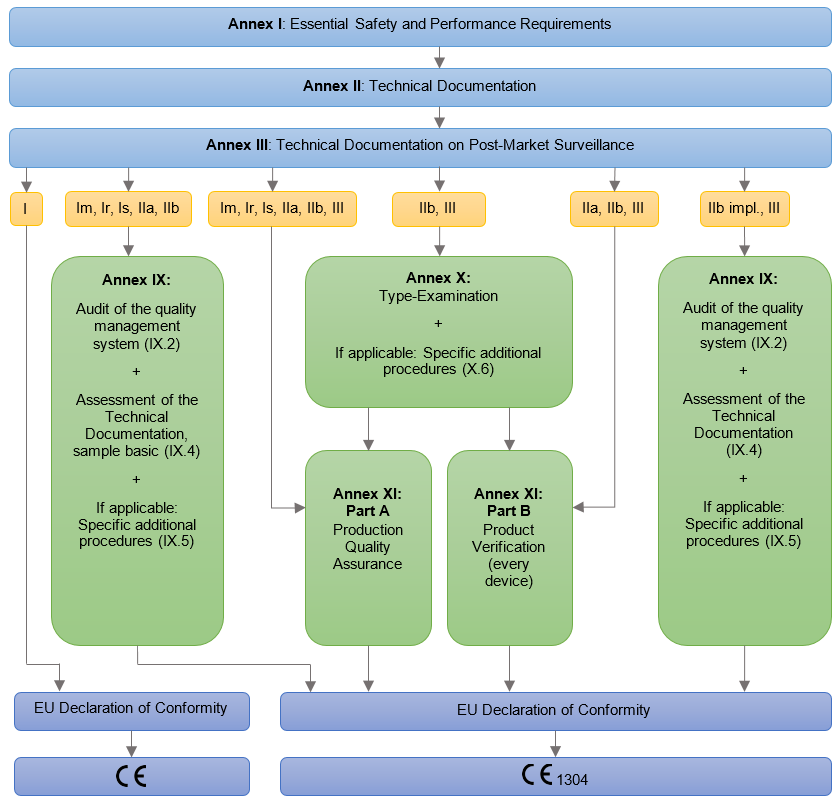
Glavni koraki v postopku pridobitve CE oznake
- Klasifikacija medicinskega pripomočka
- Vzpostavitev kontakta s priglašenim organom za medicinske pripomočke razreda Is,
- Im, Ir, IIa, IIb in III
- Izbira ustreznega postopka ocenjevanja skladnosti (odvisno od klasifikacije medicinskega pripomočka/razreda in s tem povezano stopnjo tveganja za uporabnike ter odločitev proizvajalca)
- Odločitev za vzpostavitev sistema kakovosti po ISO 13485 za proizvajalce medicinskih pripomočkov razreda Is, Im, Ir, IIa, IIb in III (z izjemo postopka ocenjevanja skladnosti po aneksu IX in XI del A)
- Izpolnjevanje bistvenih zahtev uredbe MDR
- Priprava tehnične mape z ES izjavo o skladnosti (Aneks IV MDR)
- Ocenjevanje skladnosti
- Pritrditev CE oznake na medicinske pripomočke
Tehnična dokumentacija/Tehnična mapa
Proizvajalec mora v postopku ocenjevanja skladnosti pripraviti tehnično mapo za medicinski pripomoček/skupino medicinskih pripomočkov, po kateri se preverja izpolnjevanje bistvenih zahtev medicinske uredbe. Obvezna vsebina tehnične mape je opredeljena v Aneksu II in Aneksu III uredbe MDR. Tehnično mapo ocenjuje priglašeni organ, razen v primeru medicinskih pripomočkov razreda I, ko to ni obvezno.
Preizkušanje električne in magnetne varnosti za aktivne medicinske pripomočke
Proizvajalci aktivnih medicinskih pripomočkov morajo v postopku ocenjevanja skladnosti dokazovati tudi izpolnjevanje bistvenih zahtev na področju električne in magnetne varnosti. SIQ nudi priznane in akreditirane preskusne laboratorije za preskušanje nekaterih vrst električnih medicinskih pripomočkov in opreme ter ugotavljanje elektromagnetne združljivosti. Več o tem…
Kontakt
