EU-Verordnung 2017/745 zu Medizinprodukten
Aktuelle Informationen
Nach erfolgreichem Abschluss des Benennungs- und Notifizierungsverfahrens gemäß der Verordnung (EU) 2017/745 über Medizinprodukte (MDR) wurde SIQ Ljubljana am 31. März 2022 als Benannte Stelle für Medizinprodukte benannt und in der NANDO-Datenbank der Europäischen Kommission registriert.
Im Jahr 2023 wurde die Verordnung (EU) 2023/607 verabschiedet. Sie verlängert die Gültigkeit von Zertifikaten, die gemäß der Richtlinie über Medizinprodukte (MDD) ausgestellt wurden, sowie die Übergangsfristen, innerhalb derer bestimmte Produkte, die weiterhin den Anforderungen der Richtlinie entsprechen, rechtmäßig auf dem Markt bereitgestellt werden dürfen.
Warum ein Zertifikat erwerben?
Die europäische Gesetzgebung im Bereich der Medizinprodukte (MDR) schreibt vor, dass Hersteller ihre Medizinprodukte vor dem Inverkehrbringen auf dem EU-Markt mit der CE-Kennzeichnung versehen müssen. Mit der CE-Kennzeichnung erklärt der Hersteller, dass das Medizinprodukt die Anforderungen der Europäischen Medizinprodukteverordnung erfüllt und den geltenden Sicherheits- und Leistungsanforderungen entspricht. Für Medizinprodukte der Klassen Is, Im, Ir, IIa, IIb und III ist die Einbindung einer Benannten Stelle – wie SIQ Ljubljana – in das Konformitätsbewertungsverfahren verpflichtend. Die Erfüllung der Anforderungen der europäischen Medizinprodukteverordnung wird durch die Ausstellung eines EU-Zertifikats (EU Certificate) nachgewiesen.
Zertifizierung von Medizinprodukten gemäß der EU-Verordnung 2017/745
Medizinprodukte dürfen in der EU in Verkehr gebracht werden und die CE-Kennzeichnung tragen, wenn sie den wesentlichen Anforderungen der Verordnung (MDR) entsprechen. Medizinprodukte werden nach ihrem Risikograd in die folgenden Gruppen eingeteilt:
- Klasse I – Medizinprodukte mit geringem Risiko für den Anwender;
- Klasse IIa – Medizinprodukte mit höherem Risiko für den Benutzer;
- Klasse IIb – Medizinprodukte, die ein hohes Risiko für den Anwender bedeuten und
- Klasse III – Medizinprodukte mit dem höchsten Risiko für den Benutzer.
Der Zertifizierungsprozess besteht aus einem Audit der Medizinproduktedokumentation/der technischen Unterlagen und einer Zertifizierungsbeurteilung. Es wird empfohlen, dass der Hersteller des Medizinprodukts über ein Qualitätssicherungssystem gemäß ISO 13485 verfügt. Die Beurteilung der medizinischen Vorschriften kann mit der Norm ISO 13485 kombiniert werden. Nach Erteilung des EU-Zertifikats und des Zertifikats nach ISO 13485 wird das System einmal jährlich mit regelmäßigen Audits von einzelnen Teilen des Systems überprüft und alle drei Jahre wird ein Erneuerungsaudit durchgeführt. Ein EU-Zertifikat ist gültig, solange Sie jährlich nachweisen, dass Sie die Anforderungen der ISO 13485 erfüllen.
Die wichtigsten Schritte zur Erteilung der CE-Kennzeichnung
- Klassifizierung von Medizinprodukten;
- Kontaktaufnahme mit der zuständigen Behörde für Medizinprodukte der Klassen Is,
- Im, Ir, IIa, IIb und III;
- Auswahl des geeigneten Konformitätsbewertungsverfahrens (abhängig von der Klassifikation des Medizinprodukts/der Klasse und dem damit verbundenen Risikograd für den Benutzer und der Entscheidung des Herstellers);
- Beschluss zur Einrichtung eines ISO 13485 Qualitätssystems für die Hersteller von Medizinprodukten der Klassen Is, Im, Ir, IIa, IIb und III (mit Ausnahme des Konformitätsbewertungsverfahrens in Anhang IX und XI, Teil A);
- Erfüllung von wesentlichen Anforderungen der MDR-Verordnung;
- Erstellung der technischen Unterlagen einschließlich EG-Konformitätserklärung (Anhang IV MDR);
- Konformitätsbewertung;
- Anbringung der CE-Kennzeichnung an den Medizinprodukten.
Technische Dokumentation/Technische Unterlagen
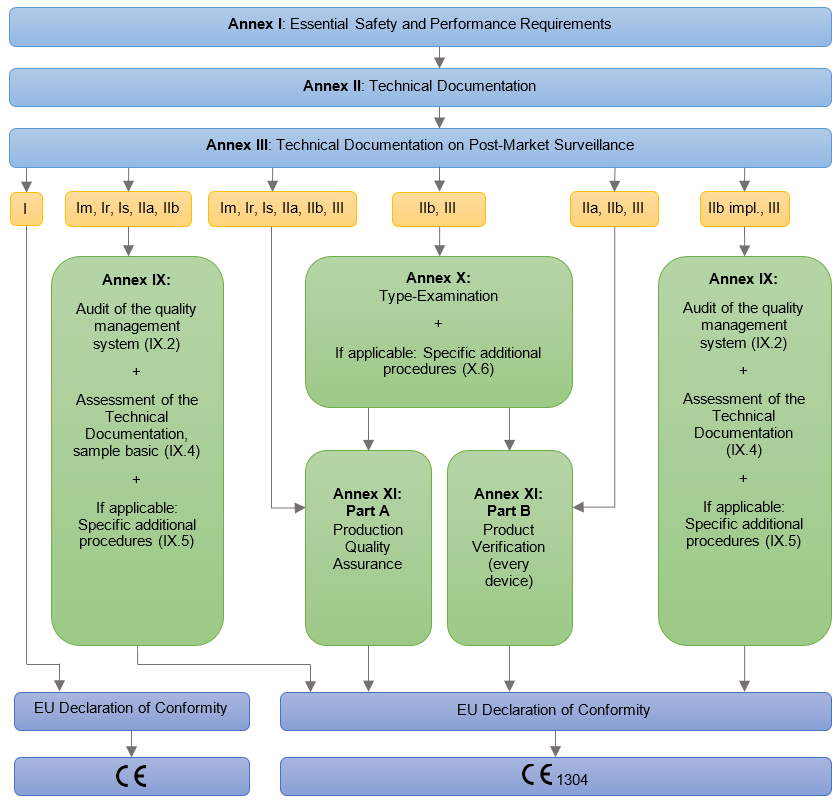
Der Hersteller muss während des Konformitätsbewertungsverfahrens technische Unterlagen für das Medizinprodukt/die Medizinproduktegruppe erstellen, wo die Einhaltung der grundlegenden Anforderungen der Medizinverordnung nachgewiesen wird. Der verbindliche Inhalt der technischen Unterlagen wird in Anhang II und Anhang III der MDR-Verordnung festgelegt. Die technischen Unterlagen werden von der zuständigen Behörde bewertet. Das gilt nicht für Medizinprodukte der Klasse I, wo dies nicht vorgeschrieben ist.
Elektrische und magnetische Sicherheitsprüfung für aktive Medizinprodukte
Hersteller von aktiven Medizinprodukten müssen im Konformitätsbewertungsverfahren auch die Einhaltung der grundlegenden Anforderungen an die elektrische und magnetische Sicherheit nachweisen. SIQ bietet anerkannte und akkreditierte Prüflabors an, um bestimmte Arten von medizinischen Elektrogeräten und Geräten zu testen und die elektromagnetische Verträglichkeit zu bestimmen.
Erfahren Sie mehr