Organization Certification, Product Testing and Certification, Training
03. August 2022


 Back to all news
Back to all news
Procedure for Certification of Medical Devices in Accordance with the Regulation (EU) 2017/745 on Medical Devices (MDR)
The path for the entry of a medical device to the market starts with an idea and leads through its production and testing to the eventual certification. It is recommended that the manufacturer contact the notified body already at the development stage of a device and obtain information on the certification procedure and the requirements that the medical device must meet.
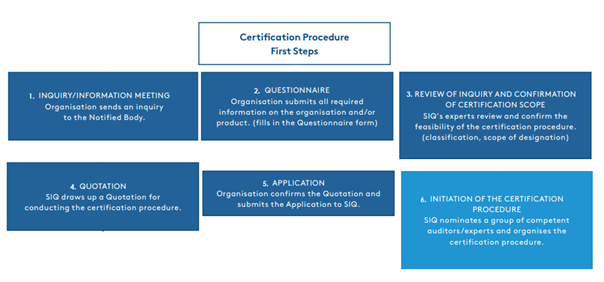
Figure 1.1.
The MDR introduces stricter and more comprehensive requirements for demonstrating the conformity of medical devices. The entire testing and certification procedure is lengthy and can take up to two years, which many manufacturers do not expect. The first steps towards starting the certification procedure, ranging from our first contact with the customer to the confirmation of the offer and the planning of the assessment and audit, are shown in Figure 1.1.
How is the certification procedure conducted?
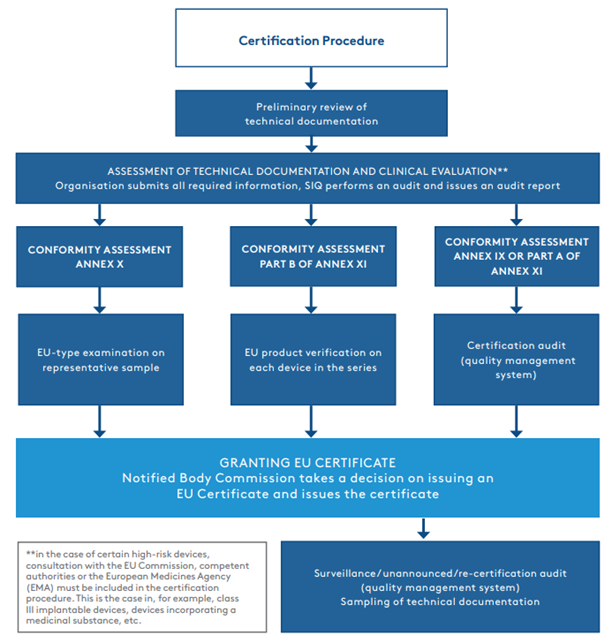
The organisation submits the technical documentation to the notified body. The latter carries out a brief review, i.e., a preliminary review of the documentation, which involves verifying the adequacy of the documentation structure and determining whether all of the required documents have been included. After confirming the adequacy and completeness of the documentation, the certification procedure can finally be initiated. The first part of the procedure is the assessment of the technical documentation and the clinical evaluation of the device. SIQ involves a range of qualified professionals, including clinical specialists and specialists with specific skills (in software, biocompatibility, sterilisation, etc.), where necessary. Once the assessment of the technical documentation and the clinical evaluation has been completed and the documentation is in compliance with the MDR, the certification procedure continues according to the chosen conformity assessment procedure (See Figure 1.2).
Most often, manufacturers decide on a conformity assessment procedure set out in Annex IX or Part A of Annex XI, which involves an audit of the established quality management system. In such cases, the certification procedure continues with a certification audit, which may be combined with an audit according to the ISO 13485:2016 standard.
Figure 1.2.
It should be noted that in the entire certification procedure, it is the assessment of the technical documentation and the clinical evaluation (part one of the assessment) that takes the longest, in some cases lasting up to one or two years.
Once all non-compliances have been eliminated by the organisation and all requirements have been met, the notified body issues a certificate (EU) that enables medical device manufacturers to enter the EU market. In the case of conformity assessment according to Annex IX or Part A of Annex XI, the notified body carries out surveillance audits of the certified quality management system once a year, followed by a re-certification audit every fifth year. During the surveillance and re-certification audits, the auditors also review the manufacturer’s reports on post-market follow-up on devices and possible complications, which are called vigilance cases. The notified body carries out an unannounced audit at least once every five years (except in the case of a Class III medical device, where the audit is carried out at least once every two years) to verify how the certificate holder is maintaining and updating its quality management system. Although the overall certification procedure in accordance with the MDR is a completely new procedure, it may be partially adapted if the organisation is already a holder of a valid EC certificate in accordance with the MDD – in the case of an
audit according to Annex IX or Part A of Annex XI, a combined MDD and MDR audit is carried out.
Find out more about the procedure.