Direktiva za medicinska sredstva (93/42/EEC)
Direktiva 93/42/EEC
Direktiva 93/42/EEC za medicinska sredstva uređuje oblast medicinskih sredstava za humanu upotrebu i obuhvata neaktivna medicinska sredstva, neaktivne implantante, sredstva za zbrinjavanje povreda, neaktivna stomatološka pomagala, medicinska sredstva vezana na izvor napajanja/aktivna medicinska sredstva, medicinska sredstva za imidžing dijagnostiku i terapiju, medicinska sredstva za nadzor/monitoring, medicinska sredstva za radio/termoterapiju, sterilna medicinska sredstva…
Zašto dobiti sertifikat
Zakonodavstvo EU (93/42/EEC) u vezi sa medicinskim sredstvima utvrđuje da proizvođači medicinskih sredstava moraju da označe svoje proizvode znakom CE, pre nego što ih ponude na tržištu EU. Time izjavljuju da je medicinsko sredstvo u skladu sa propisima EU koji uređuju oblast medicinskih sredstava i osiguravaju da je njihovo medicinsko sredstvo bezbedno i tehnički usklađeno. Kada je medicinsko sredstvo klase Is, Im, IIa, IIb i III, potrebno je da se u postupak utvrđivanja usaglašenosti medicinskog sredstva uključi i notifikaciono telo (SIQ). Usklađenost sa zahtevima propisa EU koji uređuju medicinska sredstva se iskazuje dodelom EC sertifikata (EC Certificate).
Sistem menadžmenta kvalitetom prema ISO 13485 i direktiva 93/42/EEC
ISO 13485 je harmonizovani standard kojim proizvođači medicinskih sredstava dokazuju usaglašenost sistema kvaliteta u skladu sa zahtevima direktive 93/42/EEC i, pored zahteva standarda, proizvođači moraju da uključe i posebne zahteve koje utvrđuje direktiva. Pri postupku ocenjivanja usklađenosti sa Aneksima II, V i VI, preporučujemo da proizvođač ima uspostavljen i sistem kvaliteta prema standardu ISO 13485.
Sertifikacija medicinskih sredstava prema direktivi 93/42/EEC
Medicinska sredstva mogu da se ponude na tržištu EU i da nose oznaku CE ukoliko ispunjavaju suštinske zahteve direktive za medicinska sredstva 93/42/EEC, a na tržištu Republike Srbije moraju da ispunjavaju zahteve zakona o lekovima i medicinskim sredstvima i podzakonskih akata koj iz njega proizilaze.
Medicinska sredstva se u odnosu na stepen rizika dele na:
- klasa I – medicinska sredstva s niskim stepenom rizika za korisnika,
- klasa IIa – medicinska sredstva s većim stepenom rizika za korisnika,
- klasa IIb – medicinska sredstva s visokim stepenom rizika za korisnika i
- klasa III – medicinska sredstva s najvišim stepenom rizika za korisnika.
Postupak sertifikacije se sastoji od provere dokumentacije/tehničkog fajla medicinskog sredstva i od sertifikacione provere. Preporučuje se da proizvođač medicinskih sredstava ima uspostavljen sistem kvaliteta prema standardu ISO 13485. Provera prema medicinskoj direktivi može da se kombinuje sa proverom prema standardu ISO 13485. Nakon dodele EC sertifikata i sertifikata prema standardu ISO 13485, jedanput godišnje proveravamo delovanje sistema redovnim proverama u određenim delovima sistema, a jedanput u tri godine sprovodimo resertifikacionu proveru. EC sertifikat je važeći sve dok godišnjim proverama uspevate da dokazujete da ispunjavate zahteve direktive 93/42/EEC.
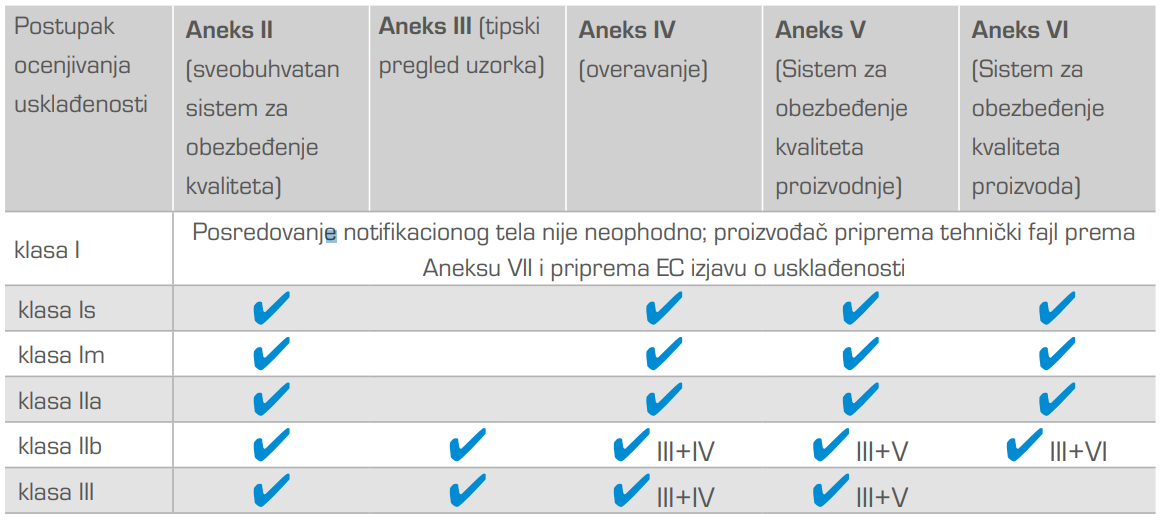
Postupci ocenjivanja usklađenosti
Postupak ocenjivanja usklađenosti medicinskog sredstva zavisi od klasifikacije medicinskog sredstva/klase i sa time povezanim stepenom rizika za korisnika, kao i od odluke proizvođača. Većina proizvođača se odlučuje za postupak ocenjivanja usklađenosti prema Aneksu V ili Aneksu II.
Postupci ocenjivanja usklađenosti medicinskih sredstava:
Glavni koraci u postupku dobijanja CE znaka
- Klasifikacija medicinskog sredstva
- Uspostavljanje kontakta sa notifikacionim telom za medicinska sredstva klase Is, Im, IIa, IIb i III
- Izbor odgovarajućeg postupka ocenjivanja usklađenosti
- Odluka o uspostavljanju sistema kvaliteta po ISO 13485 za proizvođače medicinskih sredstava klase Is, Im, IIa, IIb i III (sa izuzetkom postupka ocenjivanja usklađenosti prema aneksima III i IV)
- Ispunjavanje suštinskih zahteva direktive
- Priprema tehničkog fajla s EC izjavom o usklađenosti
- Ocenjivanje usaglašenosti
- Stavljanje CE znaka na medicinsko sredstvo
Tehnička dokumentacija/Tehnički fajl
Proizvođač mora da u postupku ocenjivanja usklađenosti pripremi tehnički fajl za medicinsko sredstvo/grupu medicinskih sredstava, po kome se proverava ispunjavanje suštinskih zahteva medicinske direktive. Obavezan sadržaj tehničkog fajla je definisan u smernici NBOG’s BPG 2009-1. Tehnički fajl ocenjuje notifikaciono telo, osim u slučaju medicinskih sredstava klase I, kada to nije obavezno.
Ispitivanje električne i magnetne bezbednosti za aktivna medicinska sredstva
Proizvođači aktivnih medicinskih sredstava u postupku ocenjivanja usaglašenosti moraju da dokažu i ispunjavanje suštinskih zahteva iz oblasti električne i magnetne bezbednosti. SIQ nudi priznate i akreditovane ispitne laboratorije za ispitivanje nekih vrsta električnih medicinskih sredstava i opreme i ocenu elektromagnetne kompatibilnosti.
Kontakt
