Uredba EU za medcinska sredstva (2017/745)
Uredba (EU) 2017/745
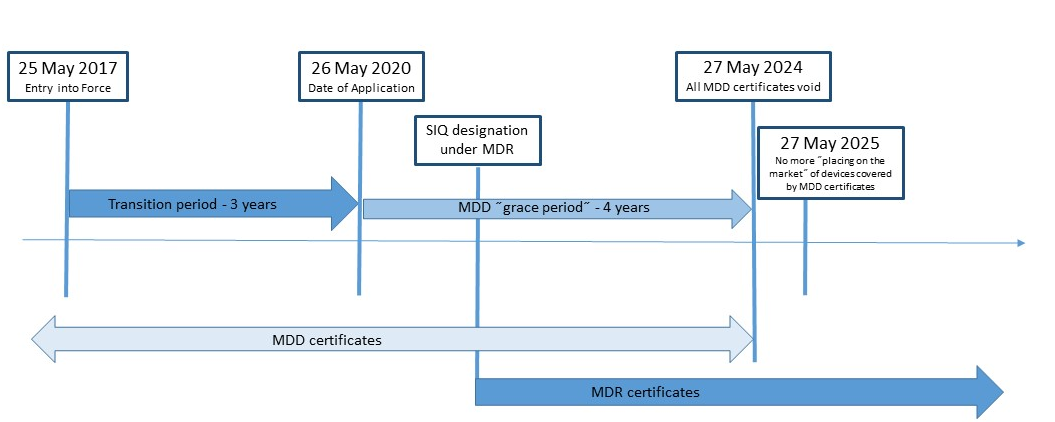
Nova Uredba (EU) za medicinska sredstva 2017/745 (MDR) objavljena je u Službenom listu Evropske unije 5.maja 2017.g i stupa na snagu 25.maja 2017.g. MDR (Medicinska regulativa) zameniće Direktivu o medicinskim sredstvima MDD 93/42/EEC i Direktivu o aktivnim implatabilnim medicinskim sredstvima (AIMD) 90/385/EGS. SIQ Ljubljana je u postupku dobijanja imenovanja po Uredbi (EU) 2017/745. Sertifikati izdati u skladu sa MDD po objavljivanju MDR (25.maj 2017.) su važeći do kraja perioda navedenog u samom sertifikatu, a koji ne prelazi pet (5) godina od datuma izdanja. Oni međutim postaće nevžeći najkasnije 27.maja 2024.g.
Zašto dobiti sertifikat?
Zakonodavstvo EU (Uredba MDR) o medicinskim sredstvima predviđa da proizvođači medicinskih sredstava svoje proizvode moraju označiti CE znakom pre nego što ih plasiraju (stave) na tržište EU. Time izjavljuju da je medicinsko sredstvo usaglašeno sa propisima EU koji uređuju (regulišu) područje medicinskih sredstava i potvrđuju da je njihovo medicinsko sredstvo bezbedno i usaglašeno sa propisima. Kada je medicinsko sredstvo klase Is, Im, Ir, IIa, IIb in III u postupak usaglašavanja sa zahtevima regulative (MDR) mora biti uključen i ovlašćeni organ (SIQ). Usaglašenost sa zahtevima propisa EU koji se odnose na medicinska sredstva dokazuju se dodeljenim EU sertfikatom (EU Certificate).
Sistem menadžmenta kvalitetom po ISO 13485 i Uredba (EU) 2017/745
ISO 13485 je harmonizovani standard kojim proizvođači medicinskih sredstava dokazuju usaglašenost sistema menadžmenta kvalitetom sa zahtevima uredbe MDR. Pored zahteva standarda proizvođači medicinskih sredstava moraju poštovati i primenjivati posebne zahteve koje propisuje uredba. Pri postupku ocenjivanja usaglašenosti po Aneksu IX i Aneksu XI u delu A, preporučuje se da proizvođač ima uspostavljen sistem menadžmenta po standardu ISO 13485.
Sertfikacija medicinskih sredstava po Uredbi (EU) 2017/745
Medicinska sredstva se mogu plasirati (staviti) na tržište EU i nositi CE oznaku ako ispunjavaju bitne zahteve uredbe (MDR). Medicinska sredstva se klasifikuju prema stepenu rizika na:
- klasa I – medicinska sredstva sa niskom stopom rizika za korisnika
- klasa IIa – medicinska sredstva sa većom stopom rizika za korisnika
- klasa IIb – medicinska sredstva sa visokom stopom rizika za korisnika i
- klasa III – medicinska sredstva sa najvišom stopom rizika za korisnika
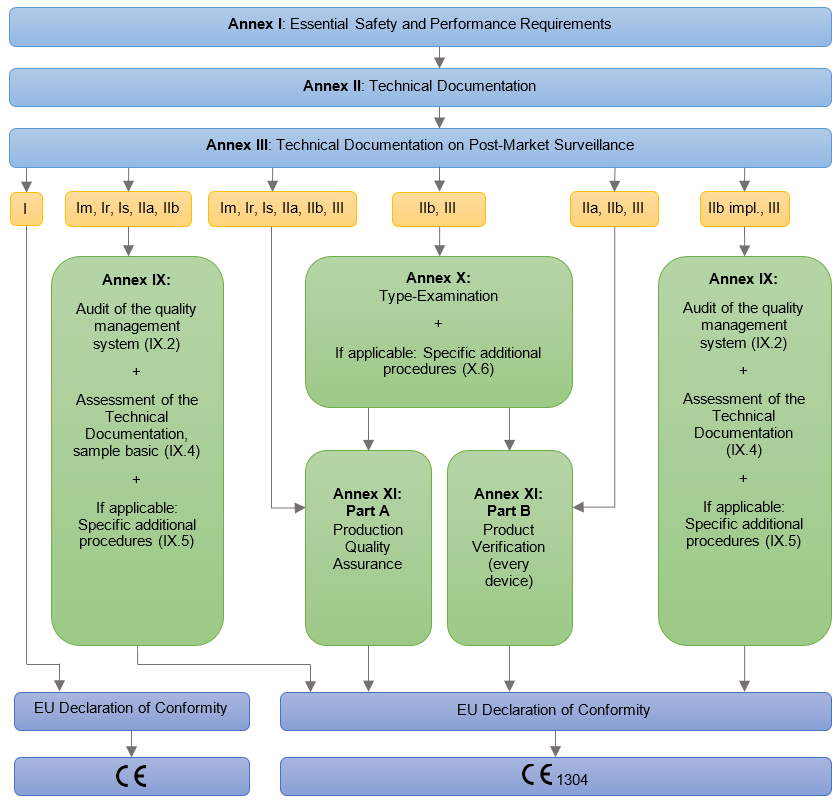
Postupak sertifikacije obuhvata proveru dokumentacije /tehničke mape (tehničkog fajla) proizvoda, medicinskog sredstva i sertifikacijske provere. Preporučeno je da proizvođač medicinskog sredstva ima uspostavljen sistem menadžmenta kvalitetom po standardu ISO 13485. Provera po medicinskoj uredbi, se može kombinovati sa proverom po standardu ISO 13485. Po dodeli EU sertifkata i sertifikata za ISO 13485 jednom godišnje redovnim nadzorom pojedinih delova sistema proveravamo funkcionisanje sistema i nakon treće godine sprovodimo resertfikacijsku (obnavljajuću) proveru za ISO 13485. EU sertfikat je važeći sve dok se godišnje dokazuje ispunjenost zaheva ISO 13485 i uredbe.
Glavni koraci u postupku dobijanja CE znaka
- Klasifikacija medicinskog sredstva
- Uspostavljanje kontakta sa ovlašćenim organom za medicinska sredstva klasa Is, Im, Ir, IIa, IIb i III
- Izbor odgovarajućeg postupka ocenjivanja usaglašenosti (zavisno od klasifikacije medicinskog sredstva / klase i s tim povezanog nivoa rizika za korisnike kao i odluke samog proizvođača)
- Odluka o uspostavljanju sistema menadžmenta kvalitetom po ISO 13485 za proizvođače medicinskih sredstava klasa Is, Im, Ir, IIa, IIb i III (osim postupka ocenjivanja usaglašenosti po aneksu X i/ili aneksu XI u delu B)
- Ispunjavanje bitnih zahteva Uredbe MDR
- Priprema tehničke mape (tehničke dokumentacije) sa EC izjavom o usaglašenosti (Aneksi II i III MDR)
- Ocenjivanje usaglašenosti
- Stavljanje CE oznake na medicinska sredstva
Tehnička mapa
Proizvođač mora u postupku ocenjivanja usaglašenosti da izradi (uradi) tehničku dokumentaciju za medicinsko sredstvo / grupu medicinskih sredstava na osnovu koje se proverava ispunjenost bitnih zahteva medicinske uredbe. Obvezni sadržaj tehničke dokumentacije je opredeljenje prema Aneksu II i Aneksu III uredbe MDR. Tehničku dokumentaciju ocenjuje ovlašćeno telo, osim u slučaju medicinskih sredstava klase I, gde to nije obavezno.
Ispitivanje električne i magnetne bezbednosti za aktivna medicinska sredstva
Proizvođači aktivnih medicinskih sredstava moraju u postupku ocenjivanja usaglašenosti dokazivati poštovanje i primenu opštih zahteva i za električnu i magnetnu bezbednost. SIQ nudi (ima i) priznate i akreditovane ispitne laboratorije za testiranje (ispitivanje) određenih vrsta električnih medicinskih uređaja i opreme te ispitivanje elektromagnetne kompatibilnosti. Više o tome…
Kontakt
